


Die Erbinformation aller Lebewesen liegt in Form von fadenförmigen Molekülen, der DNA, vor. Das DNA-Molekül besteht aus zwei langen Einzelsträngen von Nukleinsäuren, die zu einer Doppelhelix gewunden sind. Bausteine der Nukleinsäuren sind vier verschiedene Nukleotide, die jeweils aus einem Phosphatrest, einem Zucker und einer der vier Basen Adenin, Thymin, Guanin und Cytosin bestehen. Die beiden Stränge der Doppelhelix sind komplementär und werden durch physikalische Kräfte zusammengehalten. Als komplementär werden die Basenpaare aus Adenin und Thymin sowie aus Guanin und Cytosin bezeichnet. Sie liegen in den beiden Strängen einander gegenüber und sind über Wasserstoffbrücken miteinander verbunden. Aufgrund dieses Basenpaarungsmechanismus ist es möglich, aus den DNA-Einzelsträngen neue komplementäre Stränge aufzubauen.
Polymerasekettenreaktion (PCR)
Kurze, genau definierte DNA-Abschnitte (bis zu 3000 Basenpaare) können molekularbiologisch mittels der sog. Polymerasekettenreaktion (PCR) gezielt vervielfältigt (amplifiziert) werden. Hierbei wird im ersten Schritt die doppelsträngige DNA bei 95 °C in die zwei Einzelstränge aufgeschmolzen (Denaturierung). Danach wird die Temperatur auf einen Wert abgesenkt, der eine spezifische Anlagerung von Startermolekülen (Primer) an die DNA erlaubt. Die genaue Temperatur hängt von der Länge und Sequenz der Primer ab und liegt meist zwischen 55 bis 65 °C (Annealing). Ein hitzestabiles Enzym, die DNA-Polymerase, verlängert die Primer durch Anlagerung von Nukleotiden, wobei die vorhandenen Einzelstränge als Vorlage dienen und zu Doppelsträngen ergänzt werden. Diese sog. Elongation findet bei 72 °C statt.
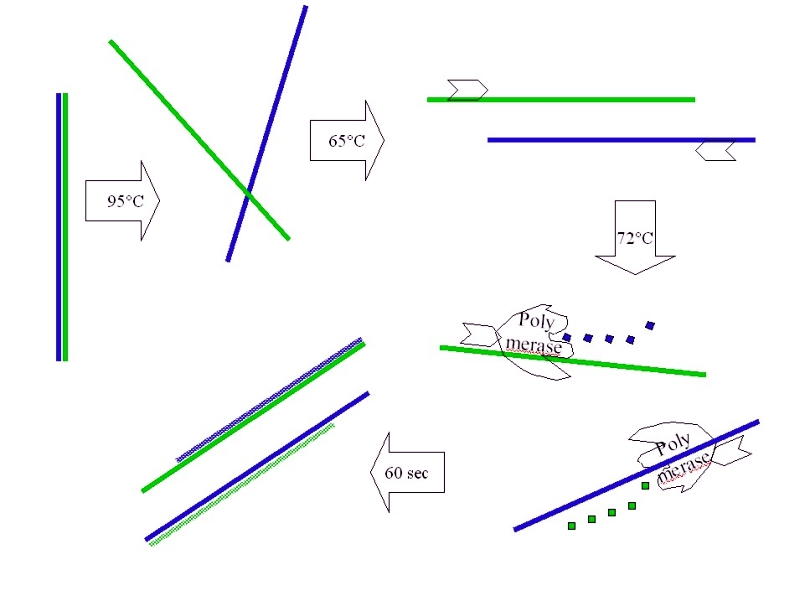 Schematische Darstellung der Einzelschritte bei der Vervielfältigung von DNA-Abschnitten (grüne und blaue Farbe) mittels PCR. Erläuterungen siehe Text.
Schematische Darstellung der Einzelschritte bei der Vervielfältigung von DNA-Abschnitten (grüne und blaue Farbe) mittels PCR. Erläuterungen siehe Text. Thermocycler zur automatisierten Durchführung der PCR-Temperaturzyklen
Thermocycler zur automatisierten Durchführung der PCR-Temperaturzyklen Diese Schritte werden 30-40 mal wiederholt. Jeder Zyklus führt zu einer Verdoppelung des DNA-Abschnitts (2-4-8-16 usw.). Nach 30 Wiederholungen können so über eine Milliarde neuer DNA-Kopien entstehen. Das wiederholte Durchlaufen der PCR-Zyklen erfolgt automatisiert in einem Thermocycler.
Gelelektrophorese
Zur Detektion der PCR-Produkte werden die amplifizierten DNA-Abschnitte im Anschluss an die PCR mittels Gelelektrophorese aufgetrennt. Die Auftrennung erfolgt in einem Trägermaterial (Agarose- oder Polyacrylamid-Gel). Da DNA negativ geladen ist, wandern die DNA-Fragmente in dem elektrischen Feld vom negativen zum positiven Pol und werden dabei nach ihrer Molekülgröße aufgetrennt. Kurze Fragmente können schneller durch die Poren des Gels wandern als größere. Nach Anfärben des Gels mit einem DNA-bindenden Farbstoff (z.B. Ethidiumbromid) fluoreszieren die DNA-Moleküle unter UV-Licht und werden als Banden sichtbar. Zur Abschätzung der Größe werden neben der zu analysierenden DNA auch Fragmente mit bekannter Größe auf das Gel aufgetragen (DNA-Leiter). Durch Vergleich mit dem Größenstandard bekannter Zusammensetzung kann die Länge der DNA-Fragmente (Anzahl der Basenpaare) abgeschätzt werden.
 Agarose-Gelelektrophorese
Agarose-Gelelektrophorese  Ergebnis der Gelelektrophorese. Die nach Molekülgröße aufgetrennten DNA-Fragmente werden im UV-Licht als Banden sichtbar.
Ergebnis der Gelelektrophorese. Die nach Molekülgröße aufgetrennten DNA-Fragmente werden im UV-Licht als Banden sichtbar. Sequenzierung
Mit Hilfe der DNA-Sequenzierung wird die Nukleotid-Abfolge in einem DNA-Molekül bestimmt. Am weitesten verbreitet ist die Didesoxymethode nach Sanger (Kettenabbruch-Synthese). Dabei wird ausgehend von einem Primer einer der beiden DNA-Stränge enzymatisch mit Hilfe einer DNA-Polymerase verlängert. Die Reaktionsansätze enthalten dabei neben den "normalen" Nukleotiden auch markierte Didesoxynukleosidtriphosphate (ddNTPs). Werden diese in den Strang eingebaut, ist eine weitere Verlängerung der DNA nicht mehr möglich. So werden DNA-Fragmente unterschiedlicher Länge gebildet, die jeweils mit einem ddNTP enden. Heutzutage werden Fluoreszenz-markierte ddNTPs verwendet, wobei jedes der vier ddNTPs mit einem anderen Farbstoff gekoppelt ist. Die entstandenen DNA-Fragmente fluoreszieren daher nach Laser-Anregung in unterschiedlichen Farben. Sie werden durch Kapillarelektrophorese aufgetrennt und mittels eines Detektors erkannt.
 Ergebnis einer Sequenzanalyse
Ergebnis einer Sequenzanalyse Die DNA-Sequenzierung wird z.B. eingesetzt bei der Artbestimmung von Organismen. Nach Amplifikation eines sog. Markergens mittels PCR wird das PCR-Produkt sequenziert und die DNA-Sequenz anschließend mit einer Referenzdatenbank verglichen.
Quantitative PCR
 Datenauswertung Real-Time PCR
Datenauswertung Real-Time PCR Die quantitativen PCR-Techniken beruhen auf dem Prinzip der herkömmlichen PCR. Bei der Real-Time PCR erfolgt die Quantifizierung über Fluoreszenzmessungen, die während des PCR-Laufs in Echtzeit (real time) stattfinden. Hierfür können DNA-bindende Fluoreszenzfarbstoffe (z.B. SYBR Green oder EVA Green) oder fluoreszenz-markierte DNA-Sonden (z.B. TaqMan-Sonden) eingesetzt werden. Die Fluoreszenz nimmt proportional zur Menge an vervielfältigter DNA zu. Der Zyklus der DNA-Vervielfältigung, bei dem die Fluoreszenz die Nachweis-Schwelle überschreitet und exponentiell anzusteigen beginnt, hängt von der ursprünglich in der Probe vorhandenen Anzahl an DNA-Stücken ab. Für die Quantifizierung ist eine Eichkurve mit Standards bekannter Kopienzahlen erforderlich.
 Datenauswertung Droplet Digital PCR
Datenauswertung Droplet Digital PCR Mit der Droplet Digital PCR (ddPCR) ist eine absolute Quantifizierung ohne Standards möglich. Der Reaktionsansatz wird auf sehr viele kleine Kompartimente (sog. Droplets) verteilt. Es entstehen Droplets, die keine Ziel-DNA enthalten und solche mit einer oder mehreren Kopien der Ziel-DNA. Die PCR-Reaktion (mit Fluoreszenzsonden) findet in jedem einzelnen Droplet statt, so dass sich am Ende ein positives oder negatives Fluoreszenzsignal für jedes Droplet ergibt. Aus dem Verhältnis von positiven zu negativen Droplets berechnet sich die Anzahl an DNA-Kopien in der Probe.